
Perceiving Crystal Symmetry#
Calculation results often specify all atoms and translation vectors, but not the space group. Here we see how to perceive the space group from a set of crystallographic coordinates.
Open a Crystal File#
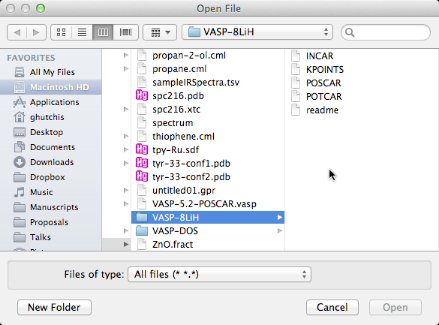
Here we open an example VASP calculation by opening the POSCAR file.
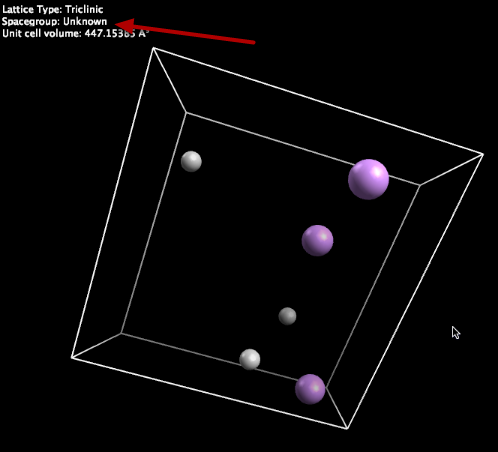
This example is triclinic, looking for Li / H structures. Note that VASP files do not specify a space group, so it is reported as “Unknown.”
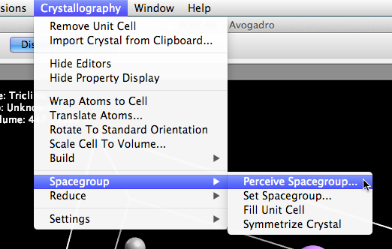
We can either set the spacegroup manually, or here, perceive the space group, using the open source spglib code.
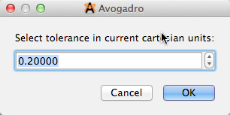
We need to set the tolerance, since some atoms may be slightly out of place in Cartesian coordinates.
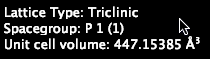
Our example VASP file isn’t very interesting – the space group is P1.
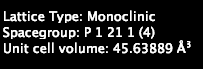
Here’s another example, where the space group is P 1 21 1.
See Also#
Building a Supercell – Build supercells from unit cells
Making a Crystal Surface Slab – Create crystal surface slabs
Building a Polymer Unit Cell – Build polymer unit cells for periodic calculations