
Finding Conformers of Molecules#
Avogadro uses Open Babel to perform conformer searches.
Start with your molecule of interest#
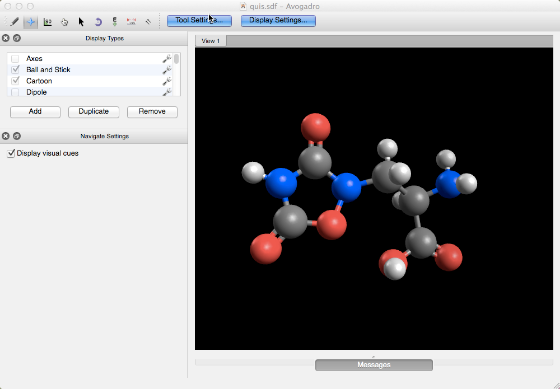
This molecule likely has several conformers and we want to be sure we have the most stable for further calculations.
Optimize Geometry#
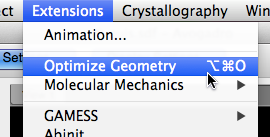
Before finding a low-energy conformer, we should make sure the current geometry is at least a near-optimal geometry. Perform an “Optimize Geometry” for a quick clean-up.
You can also use the AutoOptimize tool to perform an interactive optimization.
Configure Open Babel Force Fields#

You may want to change the force field used.
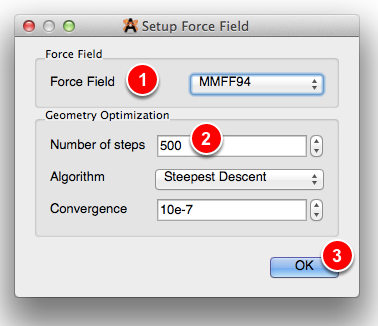
GAFF or MMFF94 are good defaults for organic-like molecules. If the molecule contains metals or is otherwise unusual, UFF is a better choice. You can also use the “Autodetect” method to set the force field based on the elements involved.
You can change the number of steps used in the conformer search in the next step.
Set the Conformer Search Options#
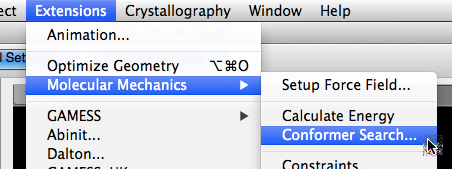
Pick a search method:
Systematic searches are exhaustive, but will always find the global minimum.
Weighted Rotor Search uses a Monte Carlo approach to learn from the energy of generated conformers. It’s a good approach for medium-to-larger molecules
Genetic Algorithm search similarly learns, and can be used for either lowest-energy or geometric diversity (RMSD).
If you pick a Weighted Search, set the number of conformers to test (e.g., 200-500).
If you pick the Genetic Algorithm search, set the options, including the scoring method.
Click “OK” to start the search.
Analyze Results#
You can view the generated conformers under Analyze ⇒ Properties ⇒ Conformer Properties...

This will bring up a window with all the generated conformers:
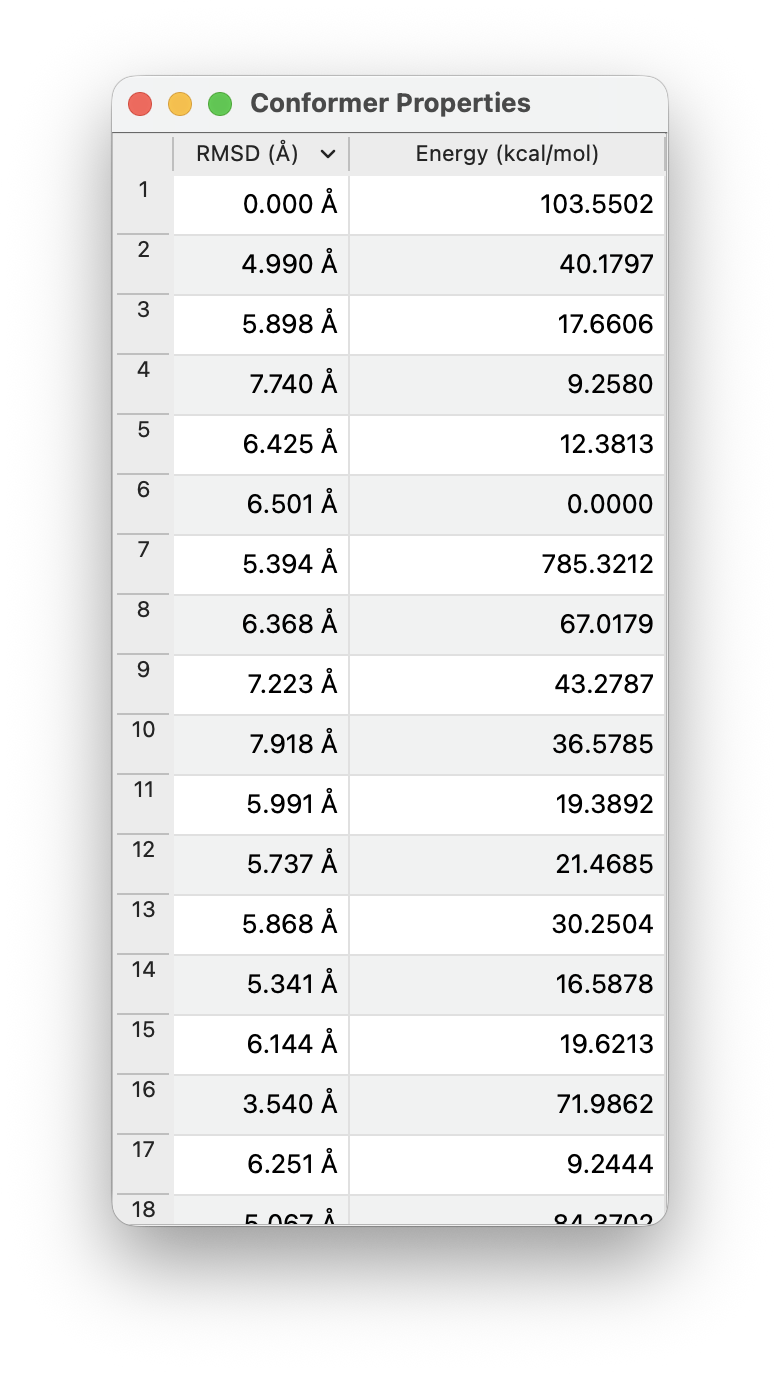
The RSMD column will indicate the root mean squared displacement of atomic coordinates (in Å) compared to the first geometry. The Energy column will indicate the relative energy compared to the lowest-energy conformer.
You can click on the two columns to sort them (e.g., to find the lowest-energy conformer).