
Geometry Constraints#
When optimizing geometries in Avogadro, sometimes you may wish to:
fix an atom at a specific position
ensure a bond or inter-atom distance remains constant
ensure an angle or dihedral remains constant
freeze the X, Y, or Z coordinates
fuse a set of atoms together while allowing them to move together
Constraints can be applied to fix (freeze) a specific selection of atoms in a molecule, as well as to fix X, Y, or Z axes, distances, and angles. For additional information see the AutoOptimize Tool and Extensions Menu documentation.
Added in version 2.0: In version 2, constraints have been reworked, offering new ways to add or remove them. A “fuse” command has also been added to enable a set of atoms to remain together (e.g. a nanoparticle or metal cluster) while optimizing other components.
Freezing Atoms#
The simplest constraint is to “freeze” an atom, or fix its Cartesian coordinates completely.
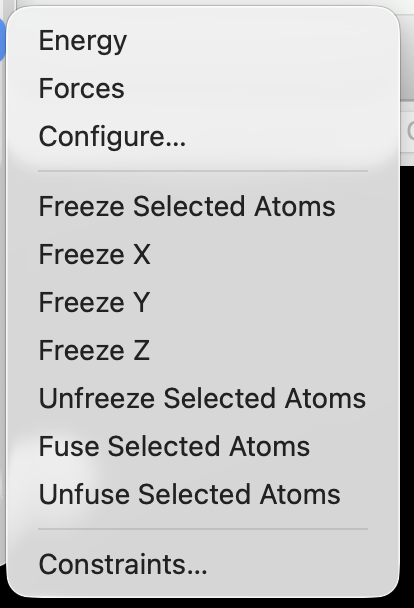
Select one or more atoms, then choose Extensions ⇒ Calculate ⇒ Freeze Selected Atoms
Freezing X, Y, or Z Coordinates#
In some cases, rather than freezing all Cartesian coordinates, you may wish to constrain only one or two (e.g., to enable the atom or atoms to move in a plane, or along a line).
Select one or more atoms, then choose Extensions ⇒ Calculate ⇒ Freeze X or Freeze Y or Freeze Z.
This can be useful, for example when optimizing a planar molecule, or adjusting a molecule above a surface.
Fusing Atoms#
If you have a set of atoms that should remain together, but can otherwise optimize (e.g., a metal cluster, ligand, or nanoparticle), you can use Extensions ⇒ Calculate ⇒ Fuse Selected Atoms.
This will create a set of distance constraints between all pairs of selected atoms, ensuring the group of atoms will retain a constant geometry even if they move together relative to the rest of the system.
Constrain a Bond or Distance (Two Atoms)#
To constrain a bond length or distance between two atoms, select the atoms with the selection tool and open the constraint dialog via Extensions ⇒ Calculate ⇒ Constraints…
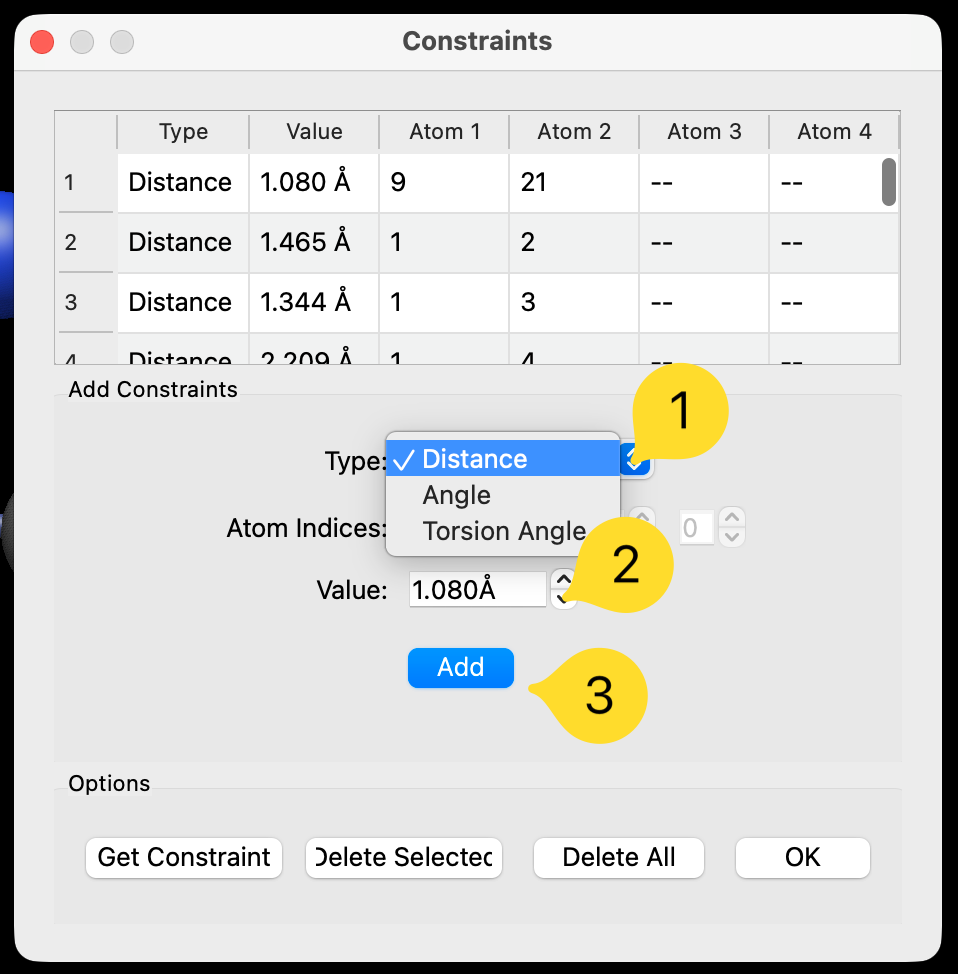
By default, the dialog will recognize that there are two selected atoms for a distance constraint, and you can adjust the type, value, and click “Add” to create the distance constraint.
Constrain an Angle (Three Atoms)#
Similarly, selecting three atoms will enable creating an angle constraint.
Constrain a Dihedral (Four Atoms)#
Selecting four atoms will by default create a torsion constraint.
Setting Constraints in Property Windows#
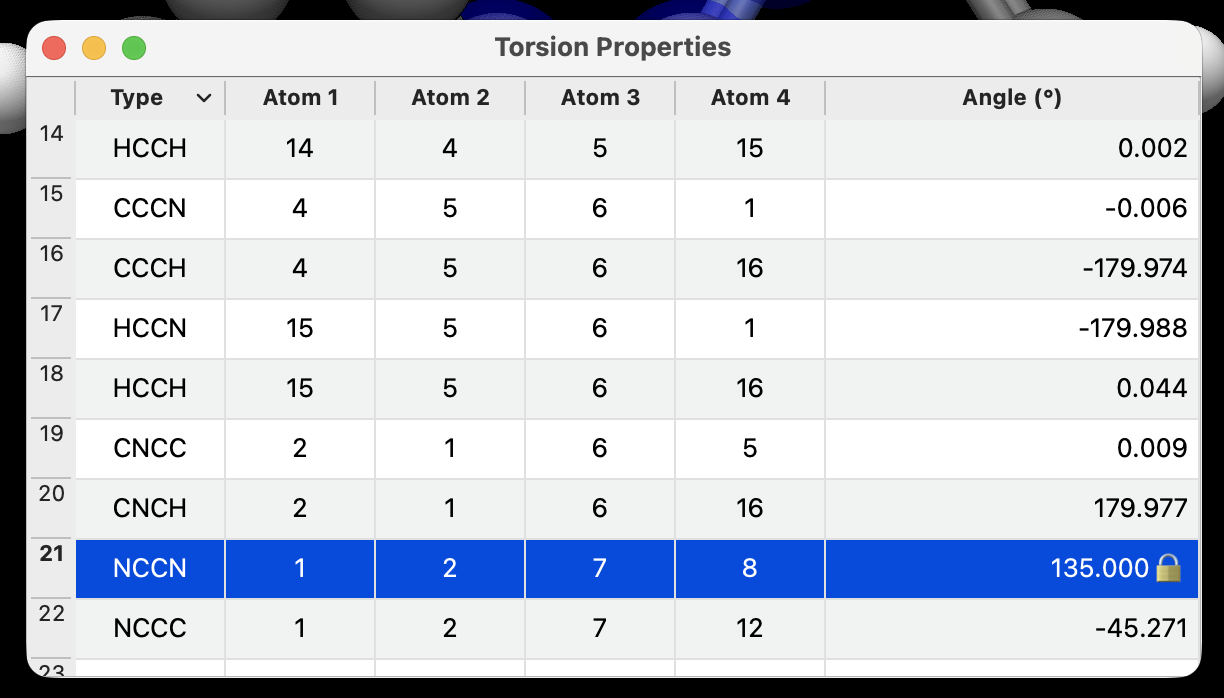
All types of geometry constraints are also shown in property windows with 🔒 icons and can be added or removed accordingly.
For example, frozen atoms will show a 🔒 for the X, Y, and Z coordinates:
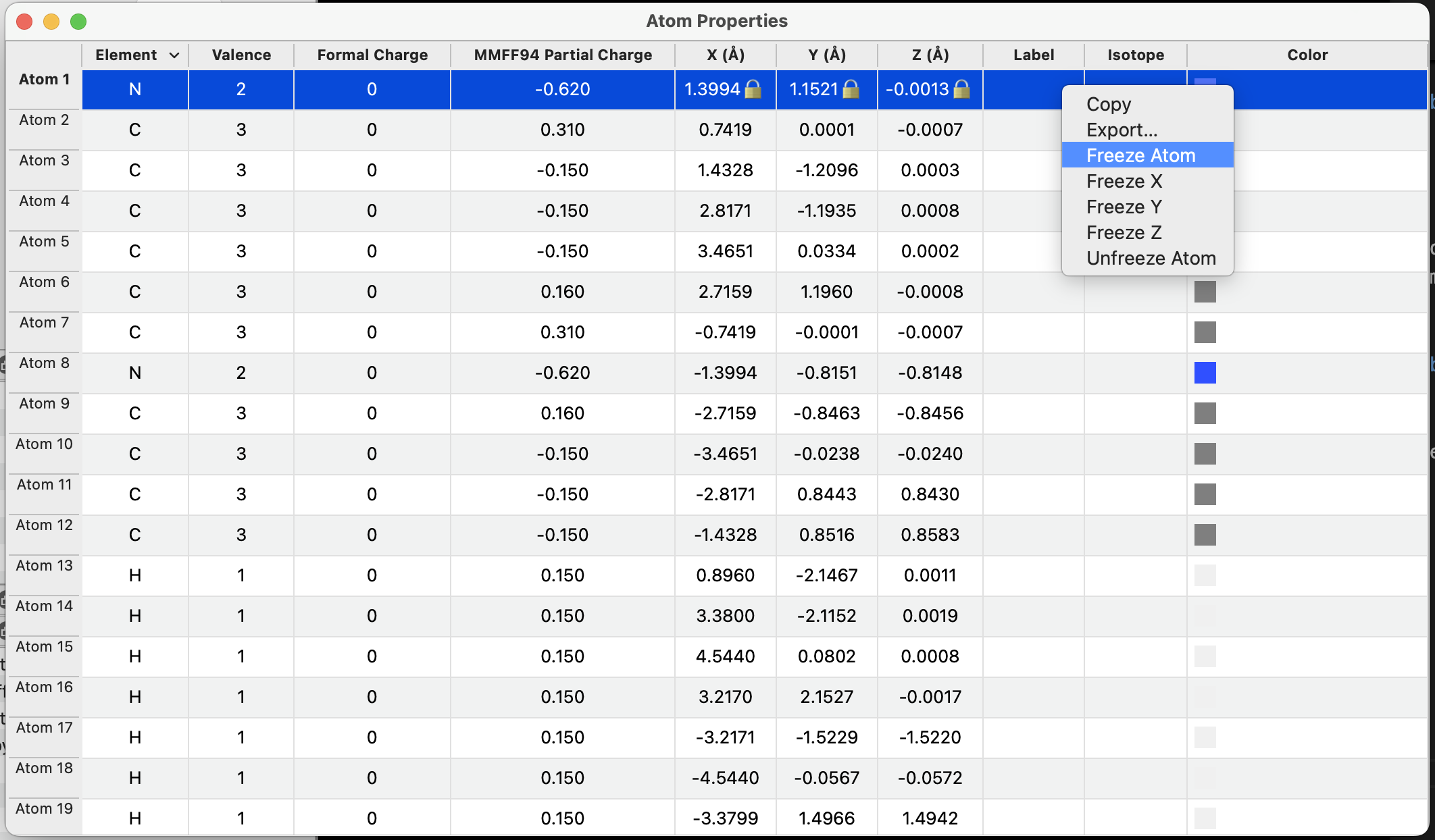
Right clicking the dialog will bring up a contextual menu to add or remove constraints on a given atom.
Similarly, distance constraints for bonds are obvious in the Bond Property dialog and the bond length is indicated with a 🔒.
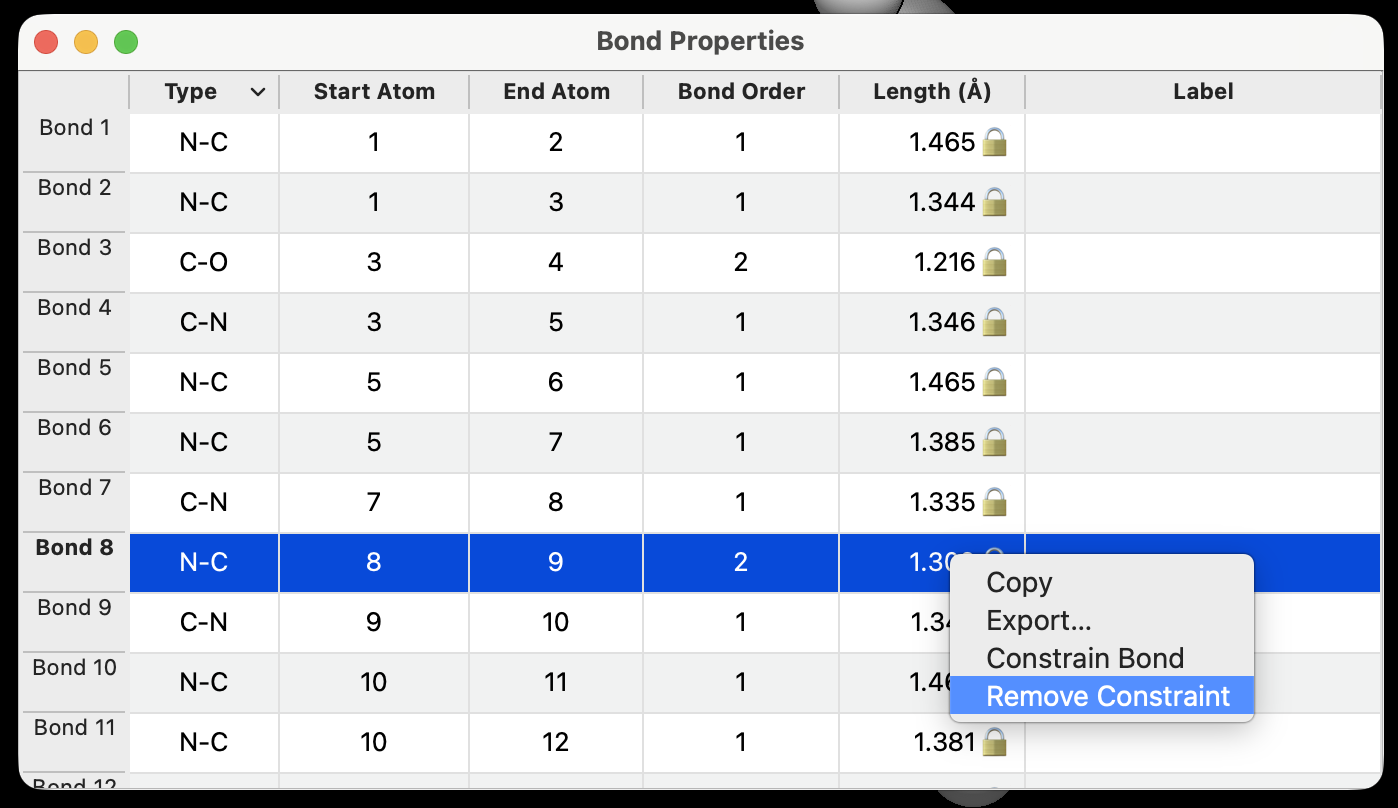
The same is true for angles or torsions:
The constraints dialog, however, enables distance constraints that are not bonded atoms, or other kinds of angles or torsions.